Research
Epithelial secretion in health and disease
The epithelial fluid and ion secretion play a central role in the maintenance of physiology of the exocrine pancreas. On the other hand, it’s damage is the trigger (such as cystic fibrosis) or booster (acute and chronic pancreatitis) of pancreatic diseases. Thus, our group investigates the detailed molecular characteristics of human pancreatic ductal secretion and possible strategies for the restoration of pancreatic ductal secretion in inflammatory pancreatic diseases. It was well known that CFTR plays a central role in maintaining pancreatic ductal secretion by mediating chloride and bicarbonate transport, and its dysfunction emerges as a key pathogenic factor in various forms of pancreatitis. However, the mechanism of these changes were not described. We showed that multiple insults—including environmental toxins, alcohol, and certain drugs—can impair CFTR expression, localization, or function in ductal epithelial cells. Cigarette smoke, particularly its heavy metal components cadmium and mercury, reduces CFTR abundance and activity in human and experimental models, leading to diminished ductal fluid and bicarbonate secretion, elevated sweat chloride, and mitochondrial dysfunction. Alcohol and its metabolites similarly disrupt CFTR folding, stability, and channel function, thereby decreasing secretion and predisposing to both acute and chronic pancreatitis. In drug-induced models, azathioprine suppresses RAC1 activity, disrupting CFTR–ezrin interactions and reducing its apical membrane localization, with consequent ion transport failure and exacerbation of pancreatitis severity. Across these diverse etiologies, sustained intracellular calcium overload and ATP depletion emerge as common downstream effects of CFTR impairment. These findings position CFTR dysfunction as a convergent mechanism in pancreatitis pathogenesis, highlighting the potential of therapeutic strategies aimed at restoring or preserving CFTR-mediated ductal secretion to prevent disease progression.
- Maléth J, Balázs A, Pallagi P, Balla Z, Kui B, Katona M, Judák L, Németh I, Kemény LV, Rakonczay Z Jr, Venglovecz V, Földesi I, Pető Z, Somorácz Á, Borka K, Perdomo D, Lukacs GL, Gray MA, Monterisi S, Zaccolo M, Sendler M, Mayerle J, Kühn JP, Lerch MM, Sahin-Tóth M, Hegyi P. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology. 2015 Feb;148(2):427-39.e16.
- Hegyi P, Wilschanski M, Muallem S, Lukacs GL, Sahin-Tóth M, Uc A, Gray MA, Rakonczay Z Jr, Maléth J. CFTR: A New Horizon in the Pathomechanism and Treatment of Pancreatitis. Rev Physiol Biochem Pharmacol. 2016;170:37-66.
- Tél B, Papp N, Varga Á, Szabó V, Görög M, Susánszki P, Crul T, Kis A, Sendstad IH, Bagyánszki M, Bódi N, Hegyi P, Maléth J, Pallagi P. Thiopurines impair the apical plasma membrane expression of CFTR in pancreatic ductal cells via RAC1 inhibition. Cell Mol Life Sci. 2023 Jan 7;80(1):31.
- Madácsy T, Varga Á, Papp N, Tél B, Pallagi P, Szabó V, Kiss A, Fanczal J, Rakonczay Z Jr, Tiszlavicz L, Rázga Z, Hohwieler M, Kleger A, Gray M, Hegyi P, Maléth J. Impaired regulation of PMCA activity by defective CFTR expression promotes epithelial cell damage in alcoholic pancreatitis and hepatitis. Cell Mol Life Sci. 2022 Apr 28;79(5):265.
- Pallagi P, Tóth E, Görög M, Venglovecz V, Madácsy T, Varga Á, Molnár T, Papp N, Szabó V, Kúthy-Sutus E, Molnár R, Ördög A, Borka K, Schnúr A, Kéri A, Kajner G, Csekő K, Ritter E, Csupor D, Helyes Z, Galbács G, Szentesi A, Czakó L, Rakonczay Z, Takács T, Maléth J, Hegyi P. Heavy metals in cigarette smoke strongly inhibit pancreatic ductal function and promote development of chronic pancreatitis. Clin Transl Med. 2024 Jun;14(6):e1733.
Epithelial signaling in health and disease
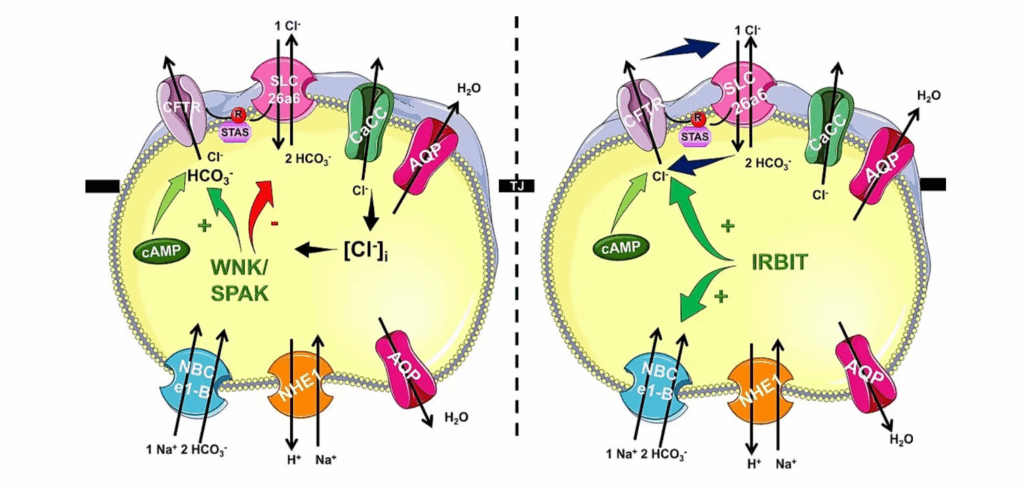
Calcium and cAMP signaling are the hallmarks of epithelial signal transduction, however overactivation of the signaling events lead to cell damage. Therefore, these signaling events are under strict control both in time (temporal regulation) and space (spatial regulation). One form of regulation is to localize these signals into subcellular microdomains, which are small regions in the cells. However, in primary, polarized epithelial cells we have only limited information about these microdomains and we don’t know how this localization affects the activity of the signaling cascades and the interactions with each other. Our research group provided novel insight into these intracellular signaling events in physiology and pathology. Our studies collectively identified Orai1-mediated calcium influx as a central driver of pancreatic ductal secretion and cell injury in pancreatitis. Structural and regulatory studies reveal that Orai1 activation and slow calcium-dependent inactivation (SCDI) are controlled by its interaction with STIM1 and the ER–plasma membrane tethering protein E-Syt1, with localization to PI(4,5)P₂-rich microdomains being essential for proper gating. Anoctamin 8 (ANO8) emerges as a critical organizer of ER–PM junctions, assembling STIM1, Orai1, and SERCA to coordinate calcium influx and prevent toxic overload. On the other hand, bile acids and ethanol synergistically activate Orai1 channels through store-operated calcium entry (SOCE), producing sustained intracellular calcium elevations that impair mitochondrial function and suppress bicarbonate and fluid secretion. Disruption of this spatial organization prolongs Orai1 activity, exacerbating calcium-driven injury. In disease models, persistent Orai1 activation by pathological stimuli leads to calcium overload, ATP depletion, and activation of cell death pathways. Pharmacologic or genetic inhibition of Orai1 markedly reduces calcium entry and protects against bile acid– and ethanol-induced ductal dysfunction. These findings underscore the importance of precise spatial and temporal regulation of Orai1-mediated calcium influx in maintaining pancreatic ductal cell homeostasis and suggest that targeting Orai1 or its regulatory complexes may be a promising strategy to mitigate pancreatitis severity.
- Madácsy T, Varga Á, Papp N, Tél B, Pallagi P, Szabó V, Kiss A, Fanczal J, Rakonczay Z Jr, Tiszlavicz L, Rázga Z, Hohwieler M, Kleger A, Gray M, Hegyi P, Maléth J. Impaired regulation of PMCA activity by defective CFTR expression promotes epithelial cell damage in alcoholic pancreatitis and hepatitis. Cell Mol Life Sci. 2022 Apr 28;79(5):265.
- Maléth J, Choi S, Muallem S, Ahuja M. Translocation between PI(4,5)P2-poor and PI(4,5)P2-rich microdomains during store depletion determines STIM1 conformation and Orai1 gating. Nat Commun. 2014 Dec 17;5:5843.
- Jha A, Chung WY, Vachel L, Maleth J, Lake S, Zhang G, Ahuja M, Muallem S. Anoctamin 8 tethers endoplasmic reticulum and plasma membrane for assembly of Ca2+ signaling complexes at the ER/PM compartment. EMBO J. 2019 Jun 17;38(12):e101452.
- Pallagi P, Görög M, Papp N, Madácsy T, Varga Á, Crul T, Szabó V, Molnár M, Dudás K, Grassalkovich A, Szederkényi E, Lázár G, Venglovecz V, Hegyi P, Maléth J. Bile acid- and ethanol-mediated activation of Orai1 damages pancreatic ductal secretion in acute pancreatitis. J Physiol. 2022 Apr;600(7):1631-1650.
- Szabó V, Csákány-Papp N, Görög M, Madácsy T, Varga Á, Kiss A, Tél B, Jójárt B, Crul T, Dudás K, Bagyánszki M, Bódi N, Ayaydin F, Jee S, Tiszlavicz L, Stauderman KA, Hebbar S, Pallagi P, Maléth J. Orai1 calcium channel inhibition prevents progression of chronic pancreatitis. JCI Insight. 2023 Jul 10;8(13):e167645
Development of advanced ex vivo disease models
Among disorders of the gastrointestinal tract diseases of the pancreas emerge due to their therapeutic difficulties and high mortality causing a major healthcare burden. Major bottlenecks of the field are the limited access to human tissue, the incomplete understanding of human pancreatic pathology and lack of human-relevant disease models. These limitations severely hinder translational pancreatic research and the development of effective therapy. To overcome these issues, novel advanced disease models with significant human relevance are crucially needed. Our group extensively uses pancreatic ductal organoids to study epithelial physiology and pathophysiology. A major breakthrough was the development of human pancreatic ductal organoids with controlled polarity by removing the extracellular matrix, which switches the orientation from apical-in to apical-out. This modification eliminates intraluminal pressure and provides direct access to the apical membrane, a long-standing limitation in studying ductal physiology. The polarity-switched organoids exhibit more stable intracellular calcium levels, improved assay sensitivity for forskolin-induced swelling, and enabled the discovery of previously unrecognized ion channels in ductal cells, including the calcium-activated chloride channel ANO1 and the epithelial sodium channel ENaC. Parallel work with mouse pancreatic ductal organoids demonstrated that these cultures closely replicate native ductal tissue in morphology, gene expression, and ion transport activity, offering a robust alternative to traditional duct isolation. Another major innovation is the establishment of human pluripotent stem cell-derived pancreatic duct-like organoids (PDLOs) that are homogeneous, polarized, and structurally, transcriptionally, proteomically, and functionally similar to in vivo ducts. These PDLOs can further mature upon transplantation into mice, forming complex ductal tissue and revealing distinct subpopulations, such as MUC1- versus CFTR-expressing ducts. Importantly, the hPSC-derived system supports precise genetic engineering, allowing the modeling of disease-relevant mutations such as KRAS, GNAS, and CDKN2A loss to recapitulate specific dysplastic and neoplastic pathways. Together, these developments bridge the gap between conventional cell lines or animal models and human pathophysiology, providing versatile, physiologically relevant platforms for basic research, drug discovery, and precision oncology.
- Molnár R, Madácsy T, Varga Á, Németh M, Katona X, Görög M, Molnár B, Fanczal J, Rakonczay Z Jr, Hegyi P, Pallagi P, Maléth J. Mouse pancreatic ductal organoid culture as a relevant model to study exocrine pancreatic ion secretion. Lab Invest. 2020 Jan;100(1):84-97.
- Breunig M, Merkle J, Wagner M, Melzer MK, Barth TFE, Engleitner T, Krumm J, Wiedenmann S, Cohrs CM, Perkhofer L, Jain G, Krüger J, Hermann PC, Schmid M, Madácsy T, Varga Á, Griger J, Azoitei N, Müller M, Wessely O, Robey PG, Heller S, Dantes Z, Reichert M, Günes C, Bolenz C, Kuhn F, Maléth J, Speier S, Liebau S, Sipos B, Kuster B, Seufferlein T, Rad R, Meier M, Hohwieler M, Kleger A. Modeling plasticity and dysplasia of pancreatic ductal organoids derived from human pluripotent stem cells. Cell Stem Cell. 2021 Jun 3;28(6):1105-1124.e19
- Varga Á, Madácsy T, Görög M, Kiss A, Susánszki P, Szabó V, Jójárt B, Dudás K, Farkas G Jr, Szederkényi E, Lázár G, Farkas A, Ayaydin F, Pallagi P, Maléth J. Human pancreatic ductal organoids with controlled polarity provide a novel ex vivo tool to study epithelial cell physiology. Cell Mol Life Sci. 2023 Jun 28;80(7):192.
- Kiss A, Farkas A, Ayaydin F, Lázár G, Varga Á, Maléth J. Human Pancreas-Derived Organoids with Controlled Polarity: Detailed Protocols and Experimental Timeline. Curr Protoc. 2024 Nov;4(11):e70045.